Reinraumklassen & Definition
DIN EN ISO 14644: Reinräume und zugehörige Reinraumbereiche
Die Internationale Organisation für Normen behandelt in der Norm 14644 das Thema Reinräume für die Halbleiter (elektronische Bauteile) sowie die Pharmaindustrie.
Reinraum Definition
Reinräume sind Umgebungen, in denen die Anzahl an Partikeln und/oder die Anzahl an Mikroorganismen auf einem konstant niedrigen Level gehalten werden. Die ISO 14644 gibt die Vorgehensweise und Anforderungen an einen Reinraum vor. Anhand von Partikelkonzentrationen wird die Luftreinheit im Reinraum klassifiziert.
Die Prüfverfahren zur Bestimmung der Luftreinheit und die Anforderungen an eine Erst-Inbetriebnahme sind vorgegeben. Außerdem wird auf die Spezifikationen von Werkbänken und Isolatoren eingegangen.
DIN EN ISO 14644-1
Mit der DIN EN ISO 14644-1 wurde Teil 1 der Norm ISO 14644 von 1999 überarbeitet und einige Reinraumanforderungen
aktualisiert. Diese technische Norm wird auch im EU-GMP-Leitfaden aufgeführt.
In der Norm wird unter anderem die Bestimmung der Reinraumklassen erläutert. Mithilfe von verschiedenen
Messpunkten in einem Raum wird die Luftreinheit bestimmt und die Reinraumklasse von den Messwerten abgeleitet.
Die Anzahl der Messpunkte im Reinraum und die statistische Auswertung dieser Werte wurden neu bestimmt. Für die
Klassifizierung der ISO 5 eines Reinraumes ist z.B. die Vorgabe für die Anzahl an 5 μm-Partikel weggefallen.
Außerdem wurde in der neuen Norm die Probeentnahme bzw. Schlauchlänge eines Messgerätes vorgegeben.
Reinraumklassen ISO
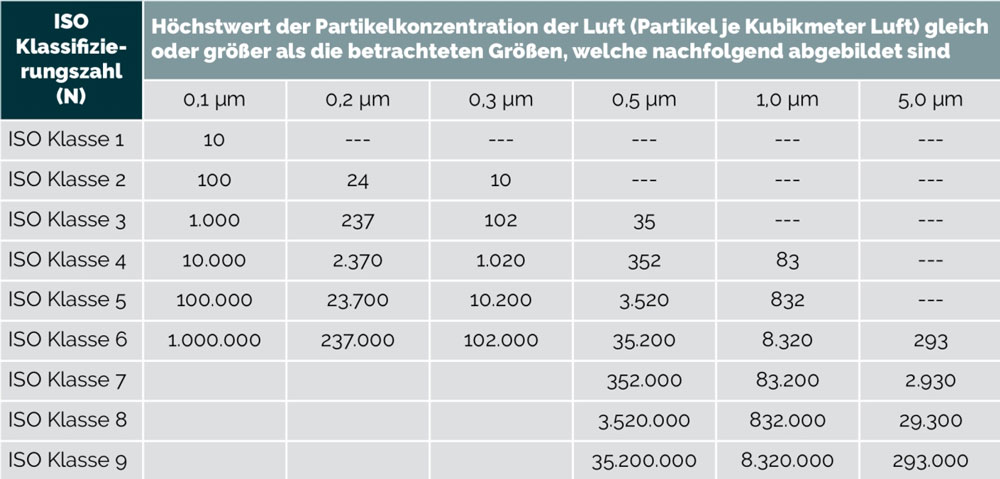
Folgende Tabelle zeigt eine Übersicht an Höchstwerten für Partikelkonzentrationen in 1 m3 Luft. Wenn ein Reinraum z.B. die ISO Klasse 6 erreichen soll, dann dürfen max. 8320 Partikel von der Größe 1μm in einem Kubikmeter Luft gemessen werden.
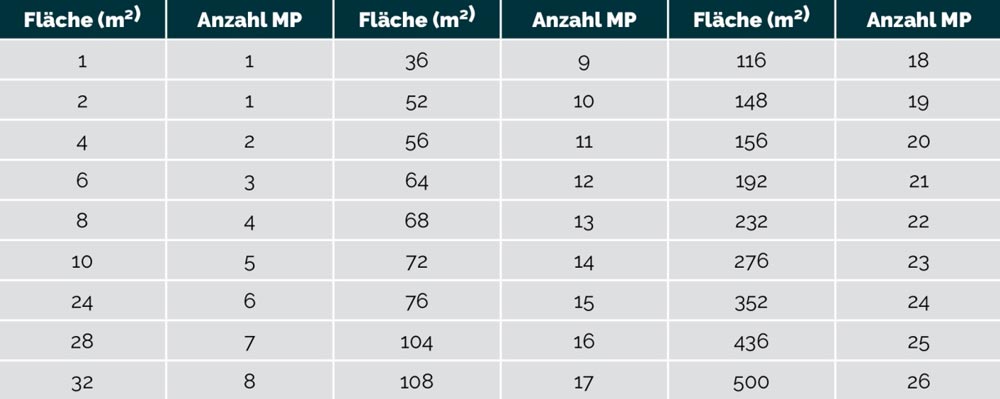
Außerdem wird in der Norm die Anzahl der Messpunkte pro Quadratmeter Reinraumfläche tabellarisch vorgegeben.
GMP-Leitfaden: „Good Manufacturing Practice“
Unter GMP versteht man die „Gute Herstellungspraxis für Arzneimittel“. Die GMP-Vorgaben werden in nationalen
sowie internationalen Regelwerken festgeschrieben. Die GMP-Regelwerke thematisieren unter anderem die
Anforderungen an die Hygiene, an die Räumlichkeiten, an die technische Ausrüstung, an Dokumentationen und
Kontrollen.
Die Regeln für die Herstellung von Arzneimitteln entwickelten sich nach verschiedenen Unglücken und Skandalen in
der Pharmaindustrie. In der Historie zwischen 1938 und 1978 entwickelte die Food and Cosmetic Act in den USA,
die WHO (Weltgesundheitsorganisation) und die FDA (Food and Drug Administration) rechtliche Grundlagen und erste
gesetzlich bindende GMP-Guidelines. In Deutschland regelt seit 1985 die Pharma-Betriebsverordnung die Gute
Herstellungspraxis. Seit 1990 besteht außerdem ein für Europa einheitlicher GMP-Leitfaden (EG GMP). In
Deutschland wird das Regelwerk „GMP“ seit 2006 durch die Durchführungsordnung Arzneimittel- und
Wirkstoffherstellungsverordnung – AMWHV geregelt.
Für einen Arzneimittelproduzenten sind die GMP-Richtlinien gültig, in dem das Produkt hergestellt wird.
Zusätzlich müssen die Regelwerke von Exportländern berücksichtigt werden. Zusammengefasst müssen folgende
Bereiche berücksichtigt werden:
- Schriftliche Arbeitsanweisungen erstellen (Standard Operating Procedure)
- Geeignete Räume und Ausrüstungen
- Qualifiziertes Personal
- Qualitätskontrollen
- Cross-Contamination-Control
- Sperrung nicht geeigneter Materialien
- Change-Control-Verfahren (Änderungsmanagement)
- Umgang mit Abweichungen (OOS-Out of Specification)
- Risk Management/Risk Assessment

Händehygiene-Anleitung als PDF
Wie Sie die Händehygiene richtig umsetzen inklusive Vorlage für einen Händehygieneplan. Jetzt E-Mail-Adresse eintragen und PDF kostenlos herunterladen.
Ich akzeptiere, dass mir gemäß den Datenschutzbestimmungen das Infomaterial zugeschickt werden darf und ich über Neuigkeiten oder Rabattaktionen informiert werde.
EU-GMP-Leitfaden der Guten Herstellungspraxis
Im Band 4 der europäischen Arzneimittelrichtlinien der europäischen Union sind die Leitlinien und Im Band 4 der Europäischen Arzneimittelrichtlinien der Europäischen Union sind die Leitlinien und Grundsätze für die Gute Herstellpraxis in der Richtlinie 2003/94/EG festgelegt.Der EU-GMP-Leitfaden wird auch als „EG-GMP-Leitfaden“ bezeichnet. Der Leitfaden besteht aus drei Teilen und derzeit 19 Anhängen, welche ständig weiterentwickelt werden. Er beinhaltet die Richtlinien zur Qualitätssicherung bei Produktion, Equipment und Reinraumumgebung in der Produktion von Arzneimitteln. Jedoch werden auch die Bereiche Wirkstoffe, Kosmetika sowie Lebens- und Futtermittel berücksichtigt. Die Anforderungen sind teilweise gesetzlich festgelegt. Die Anhänge der Leitfäden beschäftigen sich mit speziellen Anforderungen an die Herstellung von sterilen Arzneimitteln (Annex 1), mit der Herstellung von Radiopharmaka (Annex 3) sowie pflanzlichen Arzneimitteln (Annex 7).
EG-GMP-Leitfaden Teil I - GMP für Arzneimittel
Der Inhalt des EG-GMP-Leitfadens Teil 1 wird mit folgendem Auszug zusammengefasst:
„Der Inhaber einer Herstellungserlaubnis muss Arzneimittel so herstellen, dass ihre Eignung für den
vorgesehenen Gebrauch gewährleistet ist, sie den im Rahmen der Zulassung spezifizierten Anforderungen
entsprechen und die Patienten keiner Gefahr wegen unzureichender Sicherheit, Qualität oder Wirksamkeit
aussetzen.“
Weiterführend werden die Anforderungen an das Qualitätsmanagement genauer beschrieben. Darunter fallen zum
Beispiel:
- die Herstellung und Prüfung von Arzneimitteln unter gleichbleibenden Qualitätsstandards
- die Validierung kritischer Produktionsschritte
- die adäquate Schulung und Qualifizierung des Personals
Die Räumlichkeiten und Ausrüstung müssen für die Arbeitsvorgänge geeignet ausgewählt werden. Ziel bei der Wahl
der Ausrüstung ist es, das Risiko bei der Reinigung und Wartung von Kreuzkontaminationen (Verunreinigung eines
Materials oder eines Produktes mit einem anderen Material oder Produkt) und Schmutzansammlungen auf ein Minimum
zu reduzieren.
Die Dokumentation als wichtiger Teil des Qualitätsmanagements ist definiert. Die Rückverfolgbarkeit von
Chargen (eine in einem Arbeitsgang oder in einer Reihe von Arbeitsgängen gefertigte, als homogen zu erwartende
definierte Menge an Ausgangsstoff, Verpackungsmaterial oder Produkt) und Spezifikationen muss sichergestellt
sein.
Die Produktion muss der Guten Herstellungspraxis entsprechen und ist von sachkundigem Personal zu
überwachen. Dabei spielen neben der Qualitätskontrolle auch die Freigabe und Dokumentation von
Fertigerzeugnissen eine Rolle. In einem weiteren Kapitel werden gesonderte Regularien für Lohnhersteller oder
Prüflabore aufgeführt. Es ist ebenfalls die Beanstandung von Produkten und ein Rückruf geregelt. Außerdem wird
eine Selbstinspektion gefordert. Das Ziel ist dabei, fortlaufend die GMP-Vorgaben zu überwachen und ggf.
Korrekturmaßnahmen durchzuführen.
EG-GMP-Leitfaden Annex 1 - Herstellung steriler Arzneimittel
Im Annex 1 des EU-GMP-Leitfadens werden die Anforderungen an die Herstellung von sterilen Arzneimitteln in einer
spezifischeren Ausführung als im Teil1 (GMP für Arzneimittel) aufgeführt. Die Sterilität sowie weitere
Qualitätsmerkmale des Produktes sollen durch strengere Anforderungen sichergestellt werden.
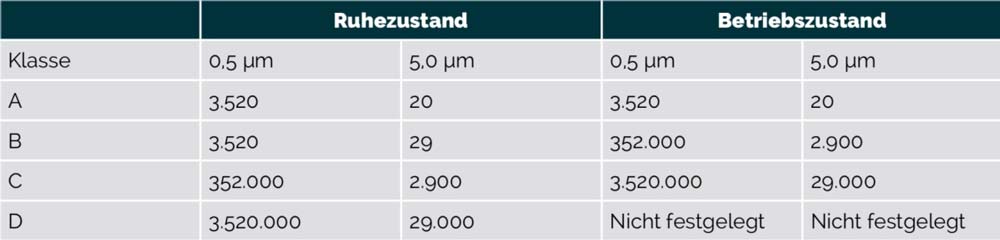
Die Produktion von sterilen Arzneimitteln dürfen in vier verschiedenen Klassen durchgeführt werden. Dabei
unterscheidet man die Reinraumklassen A, B, C und D.
Die unter Modul 3 Abschnitt C 11 aufgeführten ISO Reinraumklassen 3-8 werden mit Annex 1 wieder aufgegriffen.
Jedoch wird in diesem Regelwerk die Reinraumklassifizierung in die Klassen A-D spezifiziert.
Maximal erlaubte Partikelzahl pro m3 (gleich oder größer oder kleiner als die aufgeführte Größe)
- Die Klasse A stellt die höchste Risikostufe dar. In diesem Fertigungsbereich werden zum Beispiel offene Ampullen oder aseptische Verbindungen hergestellt.
- Die Klasse B ist die Umgebung der Reinraumklasse B
- Die Klassen C und D sind weniger kritische Bereiche bei der Herstellung von sterilen Produkten. Diese Bereiche können z.B. Zwischenstationen beim Einschleusungsprozess sein.
Des Weiteren wird im Annex 1 die Überwachung der Reinräume, z.B. durch Grenzwerte an mikrobiologischen Kontaminationen in den verschiedenen Reinraumklassen, weiter erläutert.Die Voraussetzungen zum Arbeiten in Isolatoren sind aufgeführt. Im Abschnitt „Personal“ werden die speziellen Anforderungen und Zugangsvoraussetzungen näher beschreiben. Das Personal, Reinigungs- und Wartungspersonal soll regelmäßig in den Themen sterile Herstellung, Hygiene und Grundlagen der Mikrobiologie geschult werden. Es wird zusätzlich ein hoher Standard an die persönliche Hygiene gefordert, regelmäßige gesundheitliche Untersuchungen werden empfohlen und das Tragen von Armbanduhren, Make-Up und Schmuck untersagt. Die Auswahl der Reinraumbekleidung wird detailliert aufgeführt. Außerdem werden die räumlichen Gegebenheiten wie Luftströmung zwischen den verschiedenen Bereichen genau betrachtet und geregelt. In einem weiteren Abschnitt wird die Sterilisation der hergestellten Produkte beschrieben. Nach dem Annex 1 können verschiedene Sterilisationsverfahren verwendet werden. Unter anderem eine Strahlensterilisation von hitzeempfindlichen Produkten oder eine Dampfsterilisation.
FDA - Guidance for Industry
Die Food and Drug Administration (FDA) ist in den USA die Behörde für Lebens- und Arzneimittel. Alle Unternehmen, die Produkte aus dem Bereich Nahrungsmittel, Medizin, Arzneimittel und Kosmetikartikel in die USA exportieren, müssen seit 2003 bei der FDA registriert sein. Im Jahr 1978 publizierte die FDA die ersten gesetzlichen Richtlinien (21 CFR) für die Herstellung von Pharmaprodukten. Zusammengefasst beinhalten die Richtlinien allgemeine Prinzipien der GMP Verfahrens- weisen für Arzneimittelhersteller. Die FDA veröffentlicht regelmäßig Empfehlungen für die Industrie oder Inspektoren zur Durchführung von GMP. Durch die Globalisierung der Märkte ist die FDA für deutsche Pharmaunternehmen eine ebenso wichtige wie Einflussreiche Institution, welche die Produktion von Arzneimitteln und Wirkstoffen unter GMP Vorgaben reguliert.
Literaturangaben
Bundesgesundheitsministerium: Anhang 1 zum EG-Leitfaden der Guten Herstellungspraxis (12.03.2008). Unter: https://www.bundesgesundheitsministerium.de/fileadmin/Dateien/3_Downloads/Statistiken/GKV/Bekanntmachungen/GMP-Leitfaden/Anhang-1-GMP-Leitfaden.pdf (Abruf am 27.10.2020)
Veröffentlicht am 2018-03-27 von Hygienebeauftragter Online
Stand: 2020-10-21 | Autor: Manuel Zabe